
Picture this scenario.
A horror movie where the monster is not chasing you from the shadows but rather is something that is growing inside your own flesh, in your own body.
It first begins with a harmless bump, which may resemble a minor bruise you get from bumping into something.
However, the swelling hardens instead of fading away.
Then, weeks later, you get an epiphany where the muscle isn’t healing but has mutated, turning into a solid and immovable bone.
This is not some Hollywood nightmare or a gothic curse, but rather the terrifying reality of stoneman syndrome.
Colloquially known as that term, its proper scientific term is Fibrodysplasia Ossificans Progressiva (FOP).
Being one of the rarest medical anomalies on the planet, this condition systematically replaces the soft connective tissues present in the body.
This includes muscles, tendons, and ligaments, with a secondary and permanent skeleton. With each agonizing step, the disease locks the human body into a living and breathing statue.
For centuries, this condition has blurred the line between medical mystery and psychological body horror, while trapping its victims inside a cage of their own making.
However, there is a history of profound isolation, staggering medical paradoxes and some defiant figures beneath this dread of being a ‘stoneman’.
Moreover, they refused to let this condition dictate their life, showing great resilience.
Know more about the true story of the world’s most terrifying rare disease and the extraordinary people who fought to break free from its grip.
The Science Of The ‘Gargoyle’ Curse
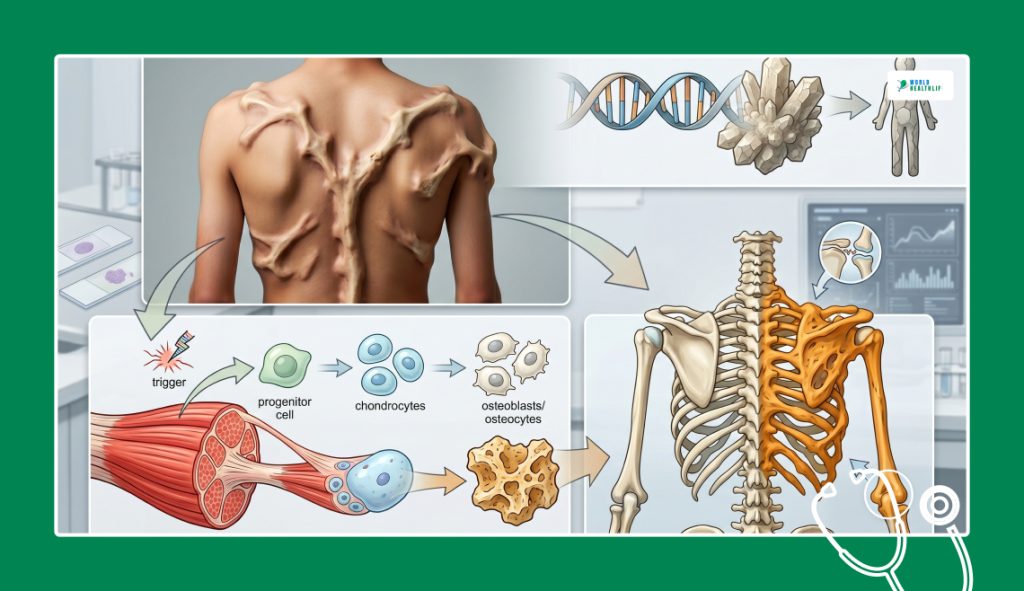
You must look at a process which is known as heterotopic ossification in medicine in order to truly have an understanding of the mechanics of stoneman syndrome.
The body deploys specialized cells to repair this specific soft tissue when a muscle or a ligament is strained.
However, in a body with Fibrodysplasia Ossificans Progressiva (FOP), this repair mechanism undergoes a catastrophic systemic failure.
The extra-skeleton bone is mistakenly constructed by the body instead of muscle or tendon tissue being regenerated.
What Is Fibrodysplasia Ossificans Progressiva (FOP)
Fibrodysplasia Ossificans Progressiva (FOP) is an extremely rare congenital genetic disorder, forcing a devastating metamorphosis within the human body.
In this condition, bone gradually and systematically replaces patients’ normal skeletal muscles and soft connective tissues, such as tendons and ligaments.
The abnormal transformation locks the affected joints permanently in place, while converting fluid movement into rigid immobility. Following this is an unrelenting path that usually becomes noticeable in early childhood.
Starting at the neck and shoulders, it steadily proceeds downward through the back, trunk, limbs and jaw. Eventually, the body fuses joints like the elbows, hips, knees, and ankles.
A pathological process known as heterotopic or extra-skeletal ossification drives Stoneman syndrome.
Heterotopic Ossification
While bone growth is normally confined to the skeleton, heterotopic ossification forces mature bone to develop in soft tissues where it should never exist.
This effectively builds a secondary skeletal cage on the outside of this natural one, where the rogue bone formation causes severe secondary complications as it infiltrates vital muscle groups.
The moment it encases the jaw, patients struggle to open their mouths, and they experience speaking and eating difficulties following severe malnutrition.
Furthermore, lung expansion is restricted, causing chronic breathing difficulties with extra-skeletal bone bridging across the rib cage.
The Genetic Glitch: What Causes Stoneman Syndrome
Variants or mutations in the ACVR1 gene cause FOP, where the gene provides instructions for making a member of a protein family called bone morphogenetic protein (BMP) type I receptors.
The protein exists in many body tissues, including skeletal muscle and cartilage. It further helps control bone and muscle growth and development.
Various studies state that variants in the ACVR1 gene cause disruptions in the mechanism responsible for controlling the receptor’s activity.
Therefore, the receptor is turned on when it normally should not be, where too much activity causes overgrowth of bone and cartilage.
The Trajectory Of Terror: Diagnosis And Prognosis
Stoneman syndrome, or FOP, manifests through the progressive transformation of soft tissues into mature bone.
The process, also called heterotopic ossification, gradually restricts mobility by locking joints and forming a second skeleton.
Sporadic mutations of the ACVR1 gene cause most reported cases, although scientists describe an autosomal dominant inheritance pattern.
Signs, Symptoms And Diagnosis
In almost all classic cases, individuals with stoneman syndrome are born with big toes that are unusually short and angled (hallux valgus).
The toe may have fused or absent joints, which is a key indicator distinguishing FOP from other musculoskeletal conditions.
Children affected with the disease are born with these congenital great toe abnormalities.
During the first decade, they develop painful soft tissue swellings, which may appear before or after trauma.
The diagnosis is primarily based on the basis of clinical symptoms, such as malformed toe and soft tissue swelling.
Targeted genetic testing for mutations in the \(ACVR1\) gene further confirms it.
Flare-Ups And The Prognosis Of Progression
The symptoms generally start appearing within the first decade of life. Oftentimes, painful tumor-like swellings or flare-ups start to develop in the soft tissues.
This starts from the neck, back and shoulders, slowly moving downwards.
These episodic flare-ups can occur spontaneously, or physical trauma, viral illnesses, or certain medical procedures (such as intramuscular injections) can trigger them.
However, doctors strictly warn against biopsies and surgeries because the trauma of an invasive procedure can provoke explosive growth of new bones in the affected area.
As the bones bridge across joints, they fuse the skeleton in place, causing permanent immobility.
The jaw is also fused, having a temporomandibular joint, leading to severe difficulties with eating and speaking.
Besides these, there are several secondary complications as well. Extra bone formation around the rib cage can cause severe restrictions in the expansion of the chest.
This causes breathing difficulties and a higher susceptibility to respiratory infections. Moreover, the condition heavily impacts the spine, frequently causing severe spinal deformities.
Statistical Isolation: 1 in 2 Million
Medical experts consider FOP to be one of the rarest genetic conditions in medicine. Characterized by the progressive transformation of soft tissues into mature bone, it effectively forms a disabling second skeleton.
Global Numbers and Patient Isolation
Stoneman syndrome is a case so rare that most doctors will never see it in their entire career. Apart from this, the condition occurs in approximately 1 in 2 million individuals worldwide.
Experts have estimated that currently there are only about 800 to 900 confirmed cases globally.
Though some estimated suggest up to 2,500 people could be affected if counting undiagnosed or misdiagnosed individuals.
Unlike many conditions, FOP does not have discriminations. The disease has shown absolutely no geographic, ethnic, racial, or gender predisposition.
The case, in fact, is so rare that most doctors might never be able to witness this in their entire career.
It is an extraordinarily rare genetic mutation, being a real-life form of body horror.
Cases Of Misdiagnosis: A Common Phenomenon In FOP
Since FOP is an ultra-rare disease, cases for misdiagnosis is common. Generally, patients are frequently misdiagnosed with aggressive cancers. This includes a diagnosis of sarcomas or aggressive forms of fibromatosis.
Consequently, invasive procedures such as biopsies or surgical interventions must be avoided at all costs. This is because trauma accelerates the runaway bone growth.
Healthcare providers lack deep familiarity with stone syndrome, and patients are usually misdiagnosed with more common conditions.
The most commonly confused diseases include cancer, fibromatosis, and lymphedema.
The Ultimate Medical Paradox
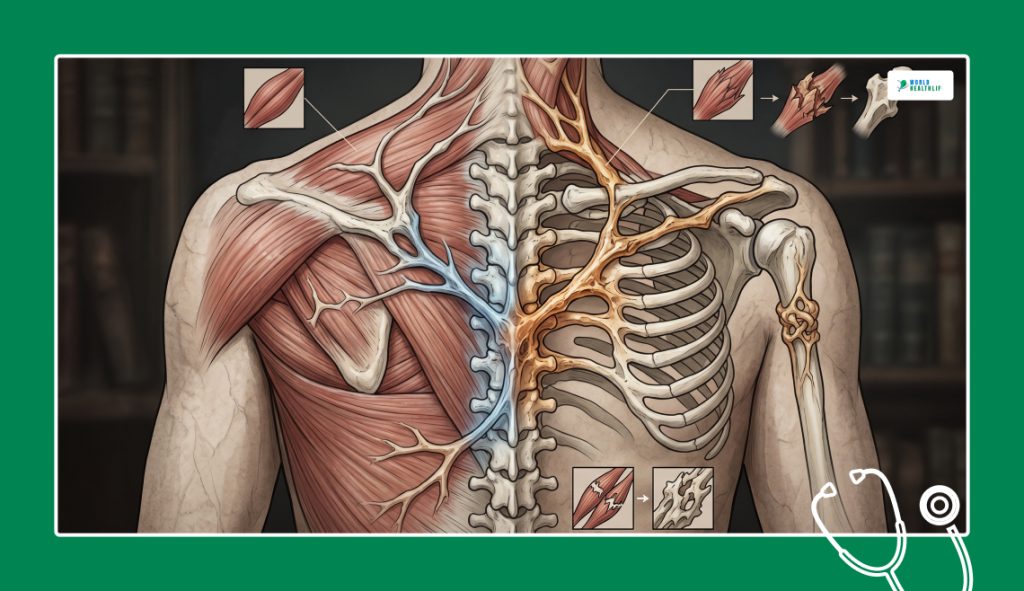
When a patient suffers from any physical obstruction or an abnormal growth restricting their movement in standard medicine, the universal protocol is surgical intervention.
This means a surgeon cuts away the restrictive tissue, frees the joint and restores the mobility of the patient.
However, for patients of FOP, there is a terrifying and devastating catch-22 situation.
Any medical attempt to surgically remove the excess bone only accelerates the disease, which makes the condition of the patient violently worse.
This cruel medical paradox is further triggered by the body’s highly sensitive and mutated immune response to trauma.
Why The Surgery Makes It Worse
The moment a surgeon’s scalpel cuts into the tissue to excise a bone bridge; the body registers the incision as a catastrophic injury.
Because a broken genetic switch permanently governs bone growth, the immune system reacts.
The reaction to the trauma of the surgery is launching an aggressive and hyper-accelerated flare-up.
Instead of healing with flexible scar tissue, the body deploys bone-building cells to the site of the wound.
As a result, the body rapidly replaces the removed bone with an even larger and completely unyielding wall of new bone.
Effect Of This Paradox
The inescapable medical paradox of stoneman syndrome strips healthcare professionals of their primary tools.
Physicians must strictly avoid standard, routine disease interventions when managing FOP.
With the most minor slip, fall, or bruise from daily life can trigger this exact same explosive ossification process.
A lack of options forces doctors into agonizing helplessness, even though they fully understand the surgery’s effects.
The Human Faces of FOP: The Story of the Pioneers
In order to look at the clinical data of FOP, we need to understand the history of the disease. To witness this is an extraordinary testament to human resilience.
Individuals suffering from FOP, who refused to let their creeping physical prisons dim their humanity, have defined the true story of the condition.
In the annals of medical history, two names from Philadelphia stand as parallel icons of this fight.
Though their lives never overlapped, fate now permanently intertwines the stories of Harry Eastlack and Carol Orzel.
Harry Eastlack: The Blueprint For A Cure
Born in Philadelphia in 1933, Harry Raymond Eastlack Jr. showed zero signs of the disease initially, other than just malformed big toes.
At just the age of five, he faced a routine childhood mishap. A car had struck him while playing, causing a fractured leg.
The trauma, however, had ignited the broken genetic switch of his FOP, while triggering a wave of explosive ossification.
Over the subsequent decades, sheets of mature bone traveled down his spine and limbs, while locking his joints into permanent angles.
By the time Harry was in his twenties, his body had completely fused into a single rigid posture. Only his eyes, tongue and lips were free to move.
Despite living in a state of constant and immense physical confinement, Harry had made a revolutionary decision.
Before he died of bronchial pneumonia in 1973, he requested that his body and medical history be donated to science.
Currently, the Mütter Museum of The College of Physicians of Philadelphia has placed his bones on display.
This offers the literal physical blueprint, which has allowed modern geneticists to map out the overall mechanics of the condition.
Carol Orzel: A Glittering Icon Of Defiance
Decades after Harry’s demise, another young woman who would pick up the mantle of advocacy came into the world in Philadelphia.
Doctors diagnosed Carol Ann Orzel with FOP as a child while she spent most of her life at a long-term care community.
Defiantly refusing to let her expanding skeleton diminish her vibrant personality, the disease also progressively claimed her physical mobility.
Furthermore, people knew her for her love of glamour, meticulous makeup routines, and a massive collection of colorful costume jewelry.
Carol did more than just survive. She went on to be a fierce and vocal educator within the FOP community.
When Carol finally ‘met’ Harry, she saw his skeleton during a temporary loan for a patient gathering.
Mesmerized by the display, she turned to her physician and made a bold statement.
With a defiant glimmer in her eye, she stated that when her time came, she wished to donate her remains to hang right next to Harry at the Museum.
She only had one specific condition. “Only if my jewelry can be displayed there too.”
Together In The Forever Home
Passing away at the age of 58 in 2018, Carol stayed true to her final wishes. The museum curators meticulously prepared her delicate remains and fulfilled the promise.
Today, visitors to the Mütter Museum can view the skeletons of Harry and Carol standing side by side in a dedicated exhibit.
Beside Carol’s completely fused, stone-like skeleton sits a vibrant display of her favorite earrings, necklaces, and brooches.
The juxtaposition is a breathtaking visual anchor for the blog post. It transforms an exhibit of severe medical pathology into an intimate portrait of identity.
Harry and Carol represent a foundational lineage of pioneers.
They shifted the global narrative of stoneman syndrome away from passive, mythological ‘body horror’ and into an era of aggressive, hopeful genetic research.
Reclaiming the Narrative From The Horror Movie
When you strip away FOP to its clinical definitions, it sounds undeniably like the plot of a cinematic nightmare.
The concept of having an unyielding secondary skeleton that locks the human inside a living statue truly brings out a primal fear.
However, the true story of FOP is more than a horror movie. People who live in it and with it are far from passive victims or medical anomalies.
As demonstrated by the pioneers, Harry and Carol, the narrative of stoneman syndrome can be defined by defiant advocacy, vibrant humanity, and an unbreakable community.
Today the collective effort is yielding an unprecedented source of hope. We are now no longer living in the era of helpless folklore pr medical mystery.
Advanced genetic engineering, targeted drug trials, and breakthrough therapies aim at silencing the faulty ACVR1 gene.
By looking past the superficial horror and understanding the profound human struggle beneath it, we bring the world closer to a day when the cage of stoneman syndrome is unlocked forever.
References:
- ACVR1 gene: MedlinePlus: https://medlineplus.gov/genetics/gene/acvr1/
- Fibrodysplasia Ossificans Progressiva: Cleveland Clinic: https://my.clevelandclinic.org/health/diseases/24476-fibrodysplasia-ossificans-progressiva
- Sekaran, L. K., Ponnuraj, N., Elangovan, V., & Sakthimohan, D. K. (2023). Fibrodysplasia Ossificans Progressiva: A rare case series. Journal of Orthopaedic Reports, 2(4), 100193. https://doi.org/10.1016/j.jorep.2023.100193
- Fibrodysplasia Ossificans Progressiva: NORD Rare Disease Report: https://rarediseases.org/rare-diseases/fibrodysplasia-ossificans-progressiva/










